I work on theoretical models of biological systems and their application to real-world data.
I am an advocate of Bayesian non-parametric models such as Dirichlet processes and Gaussian processes. I also have a background in biological sequence analysis, in particular locating transcription factor binding sites and inferring motifs that represent their binding preferences. More recently I have applied deep convolutional networks to genomic sequences to predict epigenetic marks.
In terms of applications, I mainly work with transcriptome data, most recently this has been single-cell expression data in developmental systems and also with transcription binding location data (e.g. ChIP-seq). I am interested in using these data to infer genetic regulatory networks.
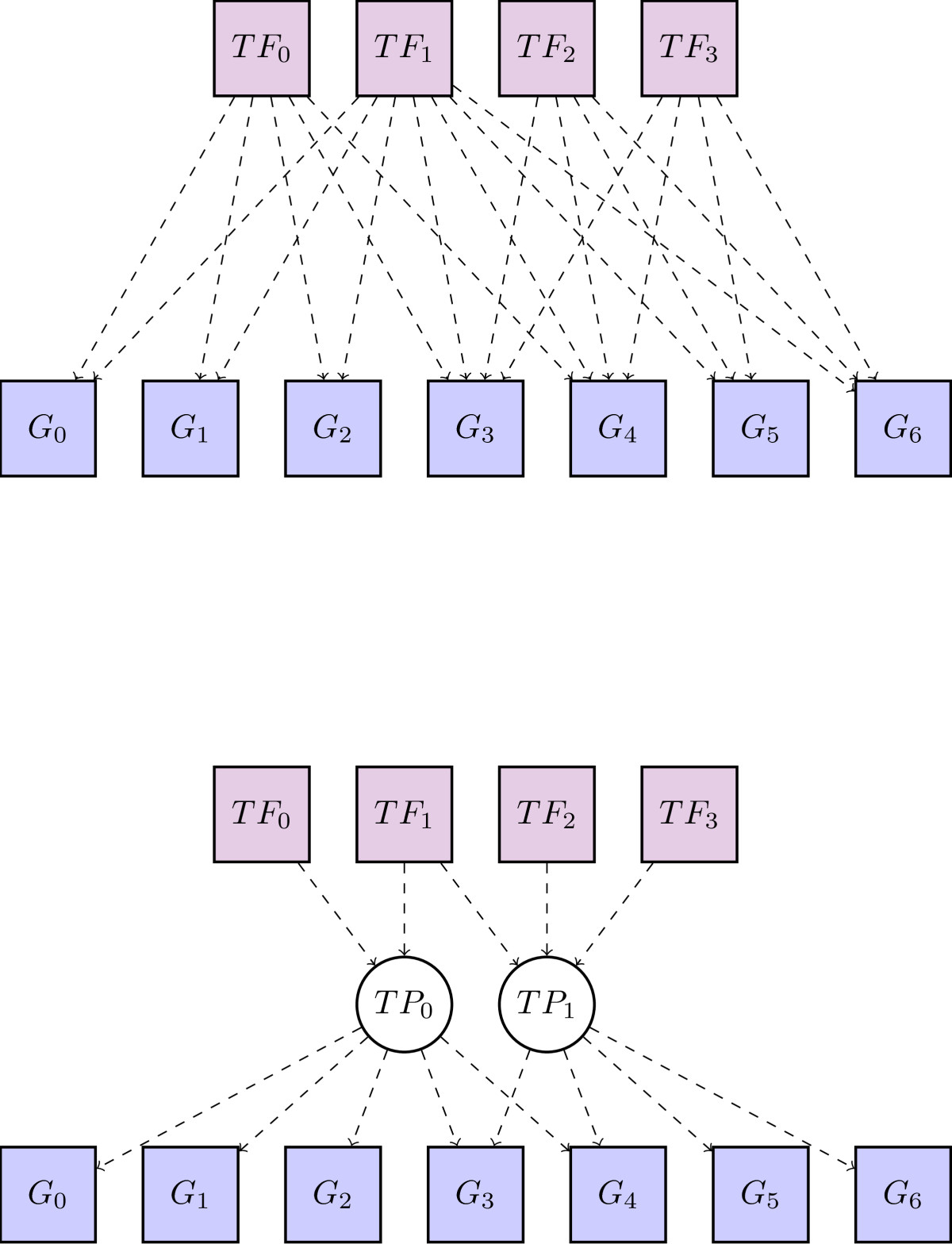
Projection layers for deep models of genomic sequence
Deep convolutional neural network models of genomic sequence capture transcription factor binding sites and other sequence features using convolutional features in their initial layer. Typically hundreds or thousands of filters are necessary for optimal predictive performance. Thus the output of the initial layer is often high-dimensional and sparse. We designed and evaluated projection layers, a method to project these internal sparse high-dimensional features into lower-dimensional dense representations. We found they improved performance and training times, suggesting they could be widely useful in this context (bioRxiv preprint).
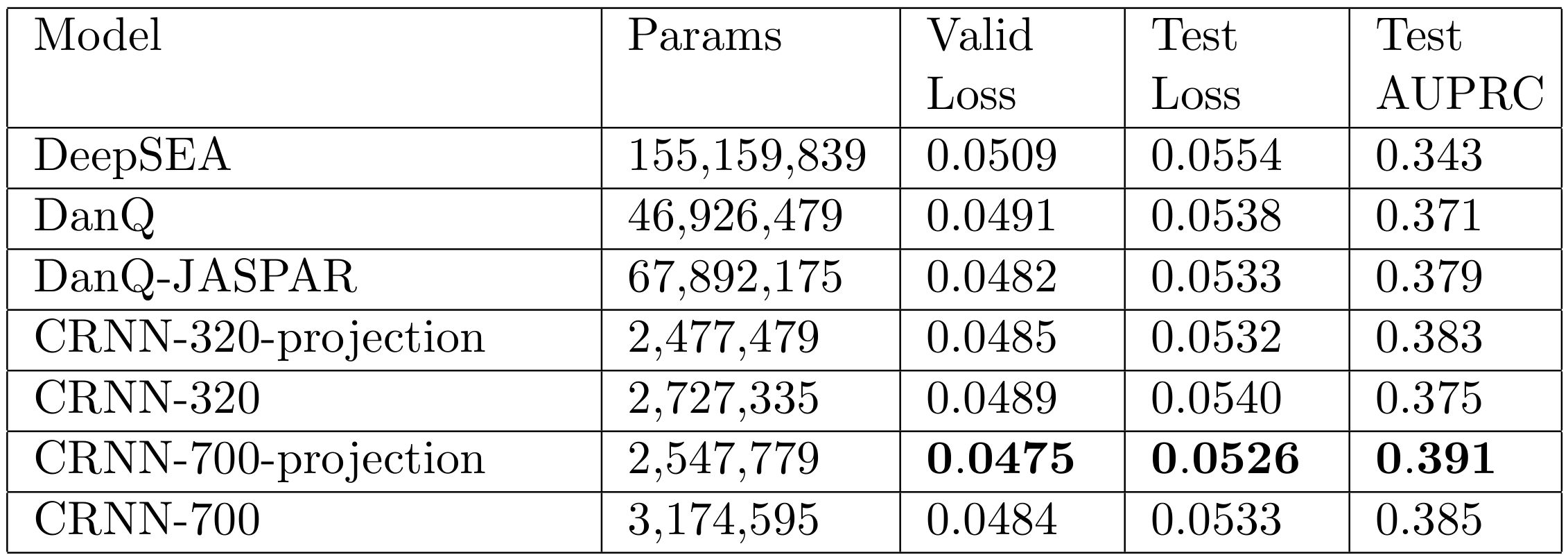
Clusternomics: integrative clustering
In genomics we frequently have information about our samples of interest from many different contexts. For example in cancer genomics, we may have gene expression, DNA methylation, micro-RNA expression and copy number variation measurements. Integrative clustering combines data from multiple contexts and suggests one overall clustering that is shared across all the contexts. Most integrative clustering models assume there is one overall clustering shared amongst the contexts and any deviations from this are more-or-less random noise. We explored the idea that there may be more complex relationships between the clusters in each context. (PLoS Comp Bio paper).
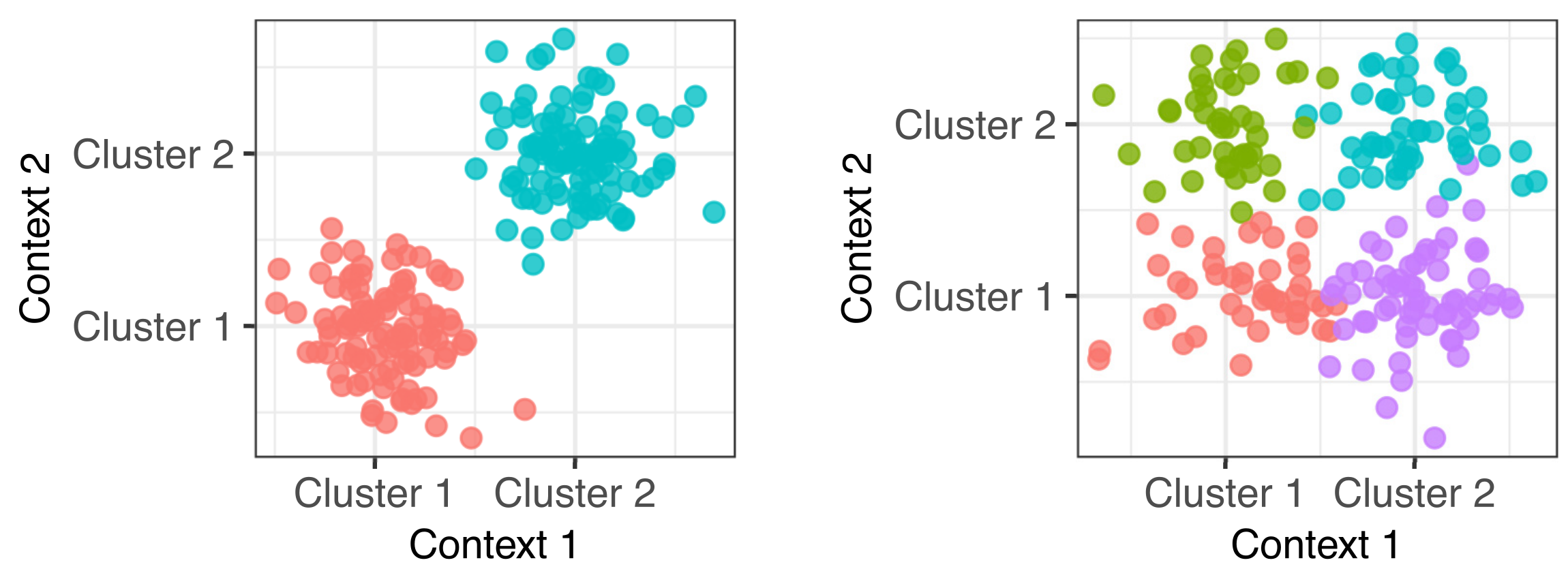
Ordering single cells in pseudotime
Many single cell experiments take samples at a handful of time points within a biological process. Unfortunately individual cells may progress through the process at different rates and this can confound any analysis of the data. In this project we modelled this effect using a latent variable Gaussian process model and inferred latent pseudotime variables to place each cell on a trajectory within the process. Downstream analyses of the data are more effective when these pseudotimes are estimated well (Bioinformatics paper).
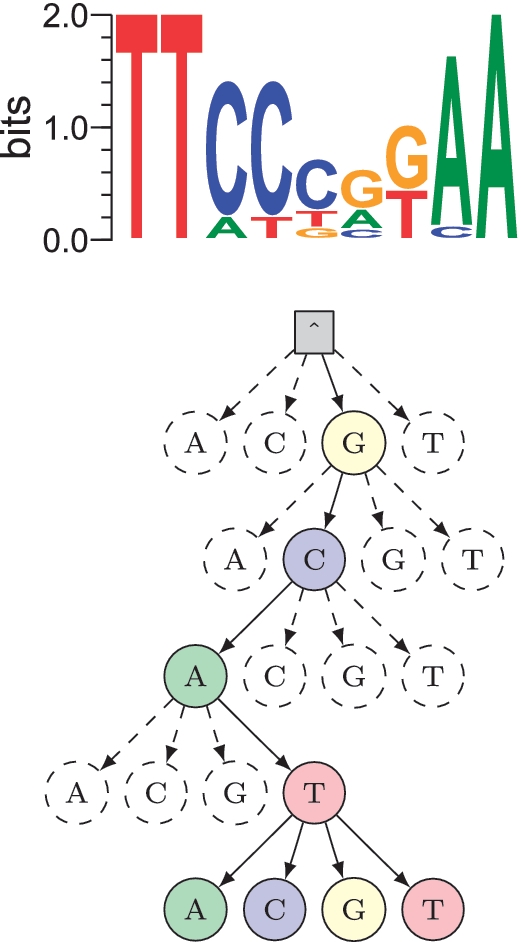
Efficient motif finding using suffix trees
Transcriptional regulation is an important mechanism that cells use to control gene expression. Unfortunately determining transcription factor binding preferences (motifs) from location data is a challenging problem. In particular experimental techniques have progressed rapidly and generate more data than state-of-the-art motif finding techniques can handle. We used a branch-and-bound approximation to significantly speed up one of the most popular motif finding methods (expectation-maximisation). This work was published in Nucleic Acids Research.
Transcriptional programs
Transcription factors do not act in isolation. Strong evidence suggests that many regulatory networks rely on combinatorial control via sets of transcription factors working together. We applied a hierarchical Dirichlet process document-topic model that is well studied in the machine learning literature to noisy data of predicted transcription factor binding sites throughout the regulatory genome. This model revealed this combinatorial structure in the form of transcriptional programs. In this application the transcriptional programs are analagous to topics in the original machine learning application (BMC Bioinformatics paper).